Aspectos gerais a serem observados nos cálculos de limites
Os Limites de Exposição Baseados em Saúde (LEBS) são a nova base para o estabelecimento de limites aceitáveis na validação de limpeza. Os cálculos de Exposição Diária Permitida (PDE) e os limites derivados para fins de validação de limpeza têm representado grande desafio analítico, podendo ser reflexo da toxicidade até então desconhecida, mas em vários outros casos, limites demasiadamente baixos tem sido calculados pelo uso equivocado do TTC de 1,5mcg/dia, aplicável para substâncias mutagênicas/genotóxicas sem dados de carcinogenicidade, para substâncias que não deveriam ser tratadas como tal.
Ademais, a aplicação exagerada de fatores de incerteza, por insegurança do avaliador (ponto de viés em estudos de avaliação de risco amplamente discutido em artigos sobre percepção de risco) ou escolha de Pontos de Partida (PODs) que se distanciam dos endpoints de toxicidade ou resposta farmacológica geralmente recomendados nas diretrizes podem ser a causa raiz de limites demasiadamente baixos e de superestimativa de risco. Além disso, algumas falhas relacionadas à apresentação dos cálculos de PDE, escolha de vias de administração ou extrapolação entre vias de administração através de um “fator de adicional” de incerteza, ou de pacientes-alvo, ao invés da consideração do uso das premissas definidas nas diretrizes de referência para o cálculo de LEBS tem sido observadas. Levantamos abaixo algumas das frequentes questões levantadas em inspeções.
1 – Devo calcular PDE para diferentes vias de administração?
Este é um tópico de dúvidas muito frequentes, e consequentemente, causa-raiz de falhas. O primeiro ponto é que o toxicologista DEVE apontar claramente para qual via está sendo calculado o PDE (ex: PDE via oral: x mg/dia), e pasme: há relatórios de cálculo de PDE em que esta informação não está clara ou sequer é apresentada. A mudança na rota do processo de fabricação com adição de um novo produto com via de administração distinta poderia ensejar ajuste e novo cálculo para outras vias de administração, mas o “PDE final” sem via de administração especificada pode acabar sendo usado para qualquer via de administração, pela ausência deste ponto no controle de mudanças.
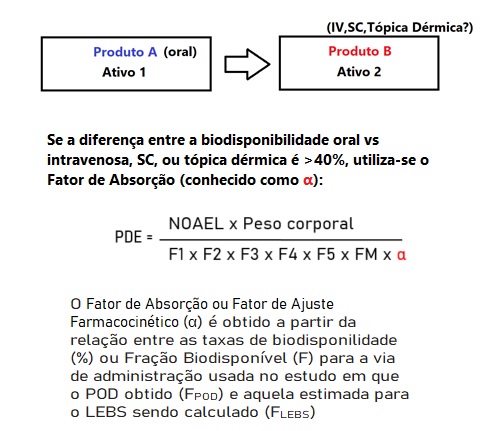
Agora, voltando ao ponto, o analista de validação de limpeza deve requisitar o valor de PDE não apenas para a via de indicação do ativo A, mas para as vias de administração do produto do lote subsequente B. Ou seja, se o ativo A é indicado para uso pela via oral, e o lote subsequente B é um produto de uso intravenoso ou tópico, o cálculo de PDE do ativo A deve para estas vias de administração do produto B devem ser incluídos na avaliação. Neste caso, um ajuste baseado nas diferenças de biodisponibilidade destas vias deve ser aplicado quando houver diferença superior a 40%. Este entendimento está previsto na diretriz PIC/s PI046-1 (2018):
“Extrapolation to other routes of administration
4.3.1 While the PDE value derived for an active substance (contaminant) generally is based on studies applying the intended clinical route of administration, a different route of administration may be applied for the active substance or medicinal product subsequently produced in the shared facility. Changing the route of administration may change the bioavailability; hence correction factors for route-to-route extrapolation should be applied if there are clear differences (e.g. > 40%) in route-specific bioavailability. As bioavailability may vary between species, the correction factors for route-to-route extrapolation should preferably be based on human data or in the case of veterinary medicinal products, data in the relevant target animal.”
2- Como deve ser aplicado o fator de correção entre vias de administração?
Dentro do possível, antes de aplicar o ajuste, deve ser realizada a busca de efeitos críticos / PODs específicos para a via de administração alvo desejada para o PDE. Por exemplo, se as vias de aministração para cálculo são oral e intravenosa, devem ser buscados valores de NOAEL para cada uma das vias específicas. Caso não haja este dado, a extrapolação entre vias é realizada com o uso do Fator de Absorção (conhecido como α), um parâmetro relativamente simples, obtido a partir das taxas de biodisponibilidade (%)específicas para cada via.
Por exemplo, caso um valor de NOAEL (Nível Sem Efeito Adverso Observado) tenha sido obtido em um estudo toxicológico realizado com a via intravenosa (mesma via de administração habitual do fármaco, com taxa de biodisponibilidade de 100% ou FPOD=1,0), e um novo cálculo de PDE para a via oral é necessário (sendo a taxa de biodisponibilidade de 50% ou FLEBS=0,5), o Fator de Absorção ou Fator de Ajuste Farmacocinético (α) será igual a 0,5. Neste caso, como raciocínio de base do cálculo, se um PDEIV fosse hipoteticamente 1mg/dia, o novo PDE obtido para a via oral seria de 2mg/dia, o que é justificável, considerando que apenas metade da dose oral seria considerada biodisponível.
3-Devo prover um cálculo de PDE para pacientes pediátricos e outro para adultos?
Não é necessário prover sempre o cálculo de um PDE pediátrico e seria impraticável fazer para todos os produtos. Todavia, este cálculo específico é recomendado quando o lote seguinte envolve uma forma farmacêutica desenhada especificamente para esta população-alvo. Temos como exemplos de produtos com apresentação tendo concentração inferior, desenhada especificamente para este público. Este entendimento está previsto na diretriz ASTM E3219 − 20, citada como referência na Nota Técnica 276/2020 da ANVISA.
“1.6 Subsequent-product HBEL values may be set for specific routes of exposure (for example, oral, inhalation, and parenteral) when necessary (for example, because of differences in bioavailability) and for specific patient populations
(for example, children) if formulations are manufactured in which one daily dose is not for the 50 kg standard adult but the dosage form is adjusted to a target population with a lower body weight.”
“The HBEL should be developed for sensitive subpopulations, which may include children. However, the HBEL is also derived for a 50 kg adult, which is not an appropriate body weight for children. Developing an HBEL for neonatal infants with an extremely low body weight is both impractical and unnecessary in most instances. Therefore, it is recommended that this adjustment be made as part of the subsequent-product HBEL in cases in which the following product is known to be administered to pediatric populations.”
“If assessing alternative populations or exposure routes (for example, infants), consult an appropriate reference (53-56). In a draft document, EMA has suggested consideration of body weight values for three pediatric populations: 0.5 kg for a prematurely born newborn, a 3.5 kg newborn, and 10 kg for a child (57).”
REFERÊNCIAS
ASTM – E3219-20: Standard Guide for Derivation of Health-Based Exposure Limits (HBELs). Current edition approved Feb. 1, 2020. Published April 2020. DOI: 10.1520/E3219-20.
EMA – EUROPEAN MEDICINES AGENCY. 2014. CHMP – Committee for Medicinal Products forHuman Use / CVMP – Committee for Medicinal Products for Veterinary Use. Guideline on setting health-basedexposure limits for use in risk identification in the manufacture of different medicinal products in sharedfacilities. EMA/CHMP/ CVMP/ SWP/169430/2012.
PIC/S. 2018. PI046-1 GUIDELINE ON SETTING HEALTH BASED EXPOSURE LIMITS FOR USE INRISK IDENTIFICATION IN THE MANUFACTURE OF DIFFERENT MEDICINAL PRODUCTS IN SHAREDFACILITIES, PIC/S, 2018, disponível em
https://www.picscheme.org/en/


